
Using whole genome optical mapping, investigators found that contrary to previous findings, the length of telomeres with viral integration was not unusually short.
Currently, it is difficult to identify the integration sites for inherited chromosomally integrated HHV-6 (ici-HHV-6) with high resolution: FISH is a challenging and laborious technique, and PCR amplification and sequencing requires previous knowledge of the chromosomal location of the integrated viral genome. For this reason, it is difficult to understand which specific loci are responsible for chromosomal integration.
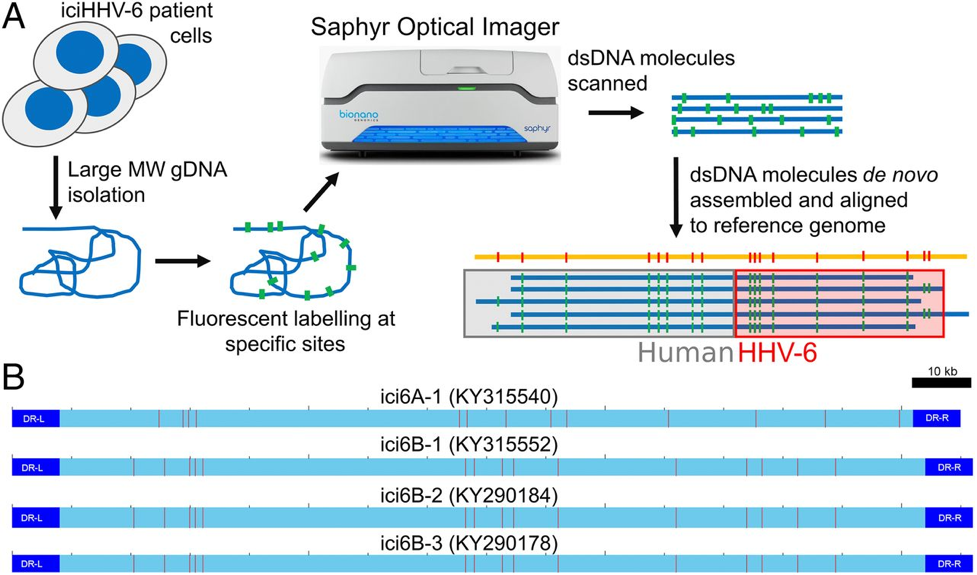
Wight et al. of the Freie Universität Berlin developed a whole-genome optical site mapping technology (Figure 1A) to obtain a high-resolution map of the integration loci within multiple patients. Direct-labeling enzyme (DLE-1) sites (Figure 1B) were created on human iciHHV-6 cell lines by covalent bonding of fluorophores and based on the predicted DLE-1 fluorophore acceptor sites in the iciHHV-6 genomes, HHV-6 integration sites were found at chromosomes 18q, 19q, Xp, and 18p. This was visually corroborated by FISH as well.
Using the optical mapping methods, the authors were also able to calculate telomere length. Contrary to previous findings, the authors found that the length of telomeres of chromosomes with viral integration was not unusually short. At the distal end of the virus genome for iciHHV-6 individuals, the telomere length ranged from 8.9 Kb to 14.7 Kb, comparable to the size of normal human telomeres. The authors suggest that the host telomere could be the main contributor for the sequences in the virus-subtelomere junction, and that the integration event likely involves activation of telomerase. Another hypothesis is that the telomere lengthening process is involved in the mechanism of genome invasion. Or, the virus could preferentially target chromosomal ends with longer telomeres.
The genome imaging approach described by the authors also has wide application for mapping transposable elements or viral integration into non-complex regions of host genomes
Read the full article: Wight 2020