
Researchers at Harvard studied how neurons responded to the presence of herpesviruses HSV1 and HHV-6, and found that these herpesviruses could rapidly induce amyloid plaque production within 24 to 48 hours. The results from this study support their earlier hypothesis that beta-amyloid (Aß) deposition and fibrillization is an innate immune response to pathogens which could protect the brain under normal circumstances.

Amyloid plaques quickly form around viral particles. Shown here are fibrils after 15 minutes, nets after 30 minutes and clumps within two hours.
Source: Eimer 2018, Neuron.
The team, based out of Massachusetts General Hospital, used transgenic AD mice that express human Aß and wild-type mice for their experiments with HSV1, and neuroglioma monolayer models as well as a 3D human stem cell-derived neural cell culture system to study HHV-6A and HHV-6B, as appropriate HHV-6 mouse models are unavailable. After injection of HSV1 into the brains of the mice, it was determined that the transgenic AD mice had significantly increased survival compared to wild-type mice, indicating higher resistance to HSV1-induced encephalitis and mortality in the transgenic mice mediated by Aß expression. In addition, host cell-associated HSV1 was lower when co-incubated in transformed H4-Aß42 and CHO-CAB media, while this effect was not seen in anti-Aß immunodepleted media. Using a modified Aß-binding ELISA, the group found that Aß oligomers bound the three herpesviruses, and the herpesvirus glycoproteins B, C, D, E, H, and G appeared to mediate the binding. They did not, however, appear to be the sole pathway responsible for Aß-targeting of the viral particles.
When the viruses were incubated with H4-Aß42 media, fibrillary structures that attached to HSV1, HHV-6A, and HHV-6B viral envelopes were rapidly generated, leading to the formation of a network of captured viral particles linked by fibrils. The viruses were found to be sequestered and neutralized by Aß fibrillization in cell culture and transgenic AD mice, as shown by virus/Aß co-localization. Extensive Aß deposits were seen in 3-week-old 3D cell neural cultures within 48 hours of infection with all three viruses- normally, Aß deposits are not generated in this type of culture until they reach 6 weeks. Similarly, in transgenic AD mice, Aß deposits do not develop until 10-12 weeks of age; however, when HSV1 was injected into 5-6-week old mice, Aß deposits were apparent in the subiculum within 48 hours following infection. Initially, Aß deposits were amorphous, but 3 weeks post-infection, Aß deposits were observed with morphologies resembling mature senile plaques found in AD brain.
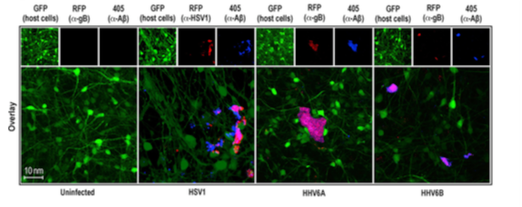
Amyloid beta deposits in pink, within 48 hours of infection with HSV1, HHV-6A and HHV-6B.
Source: Eimer 2018, Neuron.
The herpesvirus glycoproteins induced fibrillization 1-2 orders of magnitude more rapidly than that mediated by host brain glycosaminoglycans, and it may be the case that microbial sugars are the normal target for Aß in the brain, instead of the host glycosaminoglycans. Taken together, the data collected suggests that Aß is able to bind to and inhibit cellular entry of HSV1, HHV-6A, and HHV-6B, and its accumulation, which is triggered by the herpesviruses, could represent a normal, broad-spectrum innate immune response to a pathogenic challenge. Under normal conditions, this could be a protective, advantageous event that prevents viral encephalitis and acute injury; this is supported by the greater survival of HSV1-infected mice that expressed Aß compared to those that didn’t. Ultimately, however, dysfunctional accumulation of Aß may result in the accelerated development of plaques and other neuropathology. HHV-6A and –B can also affect other components of innate immunity, particularly natural killer (NK) cells, and studies have suggested that HHV-6 may contribute to Hashimoto’s thyroiditis and female infertility through modulation of NK cell activity (Rizzo 2016, Caselli 2017). The authors note that the decline of adaptive immunity and blood-brain barrier integrity as a person ages may enable HSV1 to enter the central nervous system through the olfactory route and allow increased viral replication in the brain. A similar series of events may occur for HHV-6B, and perhaps even more so for HHV-6A.
The authors note that overactivated Aß deposition may result in heightened neuroinflammation, neuropathology, and neuronal death. Consequently, it is possible that amyloidosis triggered or exacerbated by herpesvirus infection may contribute to AD.
A recently published report found a particularly strong correlation between HHV-6A and HHV-7 and AD (Readhead 2018).
Find the full paper here: Eimer 2018.