HHV-6A infection of microglia cells increases inflammatory markers and Aβ and tau expression dramatically, supporting the hypothesis that plaque development in Alzheimer’s disease may be an innate immune response to pathogens.
A group led by Roberta Rizzo, PhD at University of Ferrara in Italy set out to determine whether HHV-6A could cause the microglia to become inflamed as well as trigger Aβ deposition. They determined that TREM2, an important marker of microglial activation, and ApoE increased by 40 and 45-fold respectively, within 14 days of HHV-6A infection. Secretion of tau also occurred after 7 days, with an increasing percentage of the phosphorylated form. TREM2 interacts with ApoE, a major genetic risk factor for Alzheimer’s disease (AD), to regulate the activation of microglial cells.
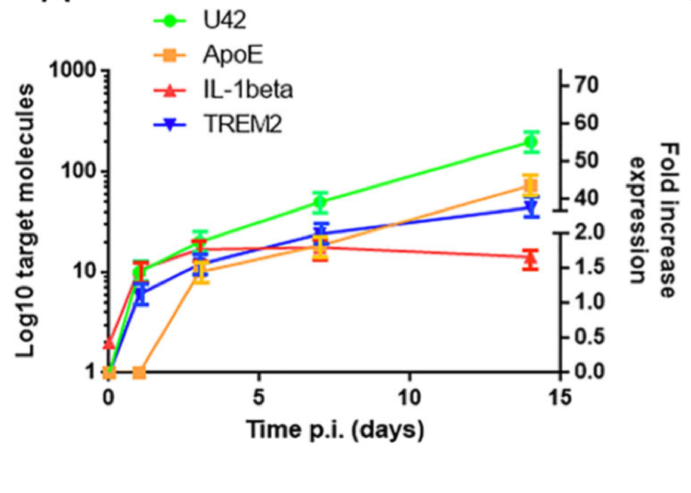
mRNA apoE, Il-1 beta and TREM2 expression over 14 days. Source: Alzheimer’s Research & Therapy
The recent realization that Aβ is an antimicrobial peptide gives credence to the infection hypothesis of AD and the authors note that while the Aβ may be beneficial in the short run, the peptide becomes progressively detrimental if the infection is chronic or reactivates repeatedly (Fulop 2018).
Of interest, the group also determined that HHV-6A infection causes migration of microglial cells to the site of infection and propose that this may be due to a paracrine effect.
Rizzo’s group previously reported that HHV-6 infection of peripheral, immune, and NK T cells induce expression of ApoE (Rizzo 2019). They also showed that HHV-6A has a significant impact on miR-155 and other key modulators in AD (Rizzo 2017).
Previous single-cell transcriptomic analysis has shown that AD brains have ApoE strongly upregulated in the microglia (Mathys 2019).
A Chinese study found that HHV-6A infection of astrocytes caused gene expression associated with several neurological diseases including AD, multiple sclerosis, mesial temporal lobe epilepsy, and glioblastomas. Differentially expressed genes included CTSS, PTX3, CHI3L1 and Mx1, all of which are genes considered targets for AD (Shao 2016).

Tau (left) and phosphorylated tau (right) expression in HHV-6 infected microglial cells.
Investigators at Steve Jacobson’s lab at NINDS/NIH also showed that HHV-6A infected astrocytes cause early increases in mRNA and protein expression of the glial glutamate transporter, EAAT-2, followed by a sustained decrease in mRNA expression and dysregulation of glutamate levels (Fotheringham 2008).
HHV-6 was first identified as a possible pathogen in AD by a British team looking for HSV1 in AD brains. While HSV1 prevalence was the same in control and diseased brains, HHV-6 was found in 70% of AD brains compared to 40% of controls (Lin 2002). HHV-6 was also found in 23% of peripheral blood leukocyte samples in patients with AD compared to 4% from controls (Carbone 2014).
Very few studies have been done in the interim, but two studies in 2018 implicating HHV-6A, HHV-7 and HSV1 as possible triggers of AD (Eimer 2018, Readhead 2018) have ignited interest in the topic. The Readhead study determined that genes involved in AD overlap with those involved in fighting viral infection and singled out HHV-6A as a key modulator of the genes involved in amyloidosis and neuronal death. HSV1 has been suggested as a trigger for AD for decades, but important findings (as reviewed by Itzhaki 2018) were largely ignored by AD researchers.
Read the full paper: Bortolotti 2019